





Abstract
Objectives
To determine the pattern of mutations of the WISP3 gene in clinically identified progressive pseudorheumatoid dysplasia (PPD) in an Indian population.
Patients and Methods
A total of 15 patients with clinical features of PPD were enrolled in this study. Genomic DNA was isolated and polymerase chain reaction performed to amplify the WISP3 gene. Screening for mutations was done by conformation-sensitive gel electrophoresis, beginning with the fifth exon and subsequently proceeding to the remaining exons. Sanger sequencing was performed for both forward and reverse strands to confirm the mutations.
Results
In all, two of the 15 patients had compound heterozygous mutations: one a nonsense mutation c.156C>A (p.C52*) in exon 2, and the other a missense mutation c.677G>T (p.G226V) in exon 4. All others were homozygous, with three bearing a nonsense mutation c.156C>A (p.C52*) in exon 2, three a missense mutation c.233G>A (p.C78Y) in exon 2, five a missense mutation c.1010G>A (p.C337Y) in exon 5, one a nonsense mutation c.348C>A (p.Y116*) in exon 3, and one with a novel deletion mutation c.593_597delATAGA (p.Y198*) in exon 4.
Conclusion
We identified a novel mutation c.593_597delATAGA (p.Y198*) in the fourth exon of the WISP3 gene. We also confirmed c.1010G>A as one of the common mutations in an Indian population with progressive pseudorheumatoid dysplasia.
Cite this article: V. Madhuri, M. Santhanam, K. Rajagopal, L. K. Sugumar, V. Balaji. WISP3 mutational analysis in Indian patients diagnosed with progressive pseudorheumatoid dysplasia and report of a novel mutation at p.Y198* Bone Joint Res 2016;5:301–306. DOI: 10.1302/2046-3758.57.2000520.
Article focus
-
The objective of this study was to screen children with clinical suspicion of progressive pseudorheumatoid dysplasia (PPD) for mutations in the WISP3 gene.
Key messages
-
Mutation c.156C>A (p.C52*) in exon 2 and mutation c.1010G>A (p.C337Y) in exon 5 of the
-
WISP3 gene are confirmed as being the most common in Indian populations.
-
A novel mutation c.593_597delATAGA (p.Y198*) has been identified, which has not been reported elsewhere.
Strengths and limitations
-
This study provides molecular validation of the clinical diagnosis of patients with PPD.
-
The study shows that WISP3 gene analysis can confirm the diagnosis of PPD in children with arthritic features and negative inflammatory markers at an early age.
-
We were unable to correlate the mutations with severity or specific clinical presentations
Introduction
Progressive pseudorheumatoid dysplasia (PPD) is a rare, autosomal recessive skeletal disorder characterised by polyarthralgia, multiple joint contractures, bulbous widening of the phalanges, prominent interphalangeal joints and short stature.1,2 Clinical symptoms of the disease manifest between the ages of three and eight years with progressive stiffness and abductor lurch.3 Juvenile rheumatoid arthritis is often mistaken for PPD because of common symptoms, widening at the interphalangeal joints and radiological features.4 In PPD, inflammatory parameters such as C-reactive protein (CRP), erythrocyte sedimentation rate (ESR) and rheumatoid factor (RF)5 are not elevated, and joint synovitis is absent.6,7 Pathology in PPD is characterised by loss of articular cartilage, resulting in early degeneration of joints and deforming arthritis, and often requiring joint replacements in the second decade of life.8
WNT1-inducible-signalling pathway protein 3 (WISP3) is a member of the CCN family. CCN is an acronym for the first three members of this family, namely cysteine-rich protein 61 (CCN1/CYR61), connective tissue growth factor (CTGF/CCN2) and nephroblastoma overexpressed (NOV/CCN3). The other members include WNT1-inducible-signaling pathway proteins (WISP1/CCN4, WISP2/CCN5, WISP3/CCN6).9 These are considered important in cell/tissue growth and differentiation.10WISP3 is a cysteine-rich multi-domain secreted protein and consists of 354 amino acids.8,11 Transient expression of wild type WISP3 gene induces collagen type II, sox9 and aggrecan expression in chondrocyte cell lines.12 Most children with PPD are missed or misdiagnosed in the early stages because of the resemblance of the early features of PPD to metabolic diseases and rheumatoid arthritis.1,5 Molecular diagnosis in the early stages will avoid unwarranted surgeries or medical treatment in these patients. So far, 60 different mutations of the WISP3 gene have been described in PPD patients, mostly in Indian, Chinese and European series.1,11,13-19 We describe the molecular abnormalities in 15 children not previously studied with phenotypic PPD who underwent mutation analysis at our centre. There have been two series reported from our country previously.14,18 In these two multicentre studies, a total of 90 patients from 79 families were screened. Mutation c.1010G>A (p.C337Y) was identified as the most common in 25 patients followed by mutation c.156C>A (p.C52*) in 21 patients.
Materials and Methods
This study was approved by the institutional review board of Christian Medical College, Vellore and followed the recommendation of the Declaration of Helsinki. Children with features of deforming arthritis, classical hand and foot changes of PPD, or suspected arthritis (but lacking in synovitis and abnormal inflammatory parameters such as elevated ESR and rheumatoid factor) were subjected to mutation analysis for WISP3.
Sample collection
Over a period of three years, peripheral blood samples were collected from 15 suspected PPD patients and their parents in EDTA-coated BD Vacutainer tubes, after obtaining informed consent. Genomic DNA was extracted using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The blood samples were lysed using proteinase K and each lysate was transferred to a spin column. Nucleic acids were bound in the spin column and the remaining contaminants were removed while washing with buffers. Finally, 200μl of AE buffer was used to elute the DNA from the column.
Mutation analysis
All primers for the exons of WISP3 were designed using Primer-BLAST software20 and obtained from Shrimpex Biotech Services Pvt Ltd (Chennai, India) (please see Table i in the supplementary material online). Genomic DNA was amplified by Polymerase Chain Reaction (PCR) using the Eppendorf Mastercycler gradient. The PCR programme consisted of an initial denaturation at 96°C for five minutes, 35 cycles of denaturation at 96°C for one minute, primer annealing (temperature varies with exon, please refer to Table i) for 30 seconds, extension for 30 seconds at 72°C and a final extension at 72°C for ten minutes. Amplified DNA was confirmed by using 2% agarose gel.
Conformation-Sensitive Gel Electrophoresis
Conformation-Sensitive Gel Electrophoresis (CSGE) was undertaken for heteroduplex analysis. To generate duplexes, 4 µl of patient and normal PCR products were mixed and denatured at 95°C for five minutes and allowed to renature at 55°C for 30 minutes. The duplexes were then electrophoresed using 10% polyacrylamide gel. The PCR product which formed a heteroduplex and showed an abnormal band formation was subsequently confirmed by Sanger’s direct sequencing method using a 3130 Genetic Analyzer (Applied Biosystems, Foster City, California).
Computation analysis
All patient sequences were analysed using the BLAST software (basic local alignment search tool) (http://blast.ncbi.nlm.nih.gov/Blast.cgi).20 Mutations were detected by comparing patient sequences with normal sequences (NG_011748.1).
Results
In total, 15 children (eight females and seven males) were diagnosed clinically as having PPD during the study period of three years. In all of our patients, the diagnosis of PPD was supported by classical clinical and radiological features. The average age at onset of symptoms was 4.4 years (1.5 to 8) and at diagnosis, 11 years (9 to 14). Initial presentation in most children was stiffness of hands, feet and restricted motion (Fig. 1). One patient presented with deformity at the elbow, one at the knee and another one at the hip. At the time of diagnosis the most common symptom was locomotor difficulty, with almost half of the patients having a painful gait and one child completely unable to walk. Radiologically, irregular narrowing of the joint space was seen in the hips (14 patients), knees (11 patients) and ankles (12 patients) in the lower limbs, while in the upper limbs the hands and elbows were affected in 13 children (Figs 2a, 2b and 2c). Platyspondyly was present in all patients but scoliosis4 and spondylolisthesis4 were detected in fewer children (Fig. 2d). In all, four patients had undergone previous surgeries for deformity correction with a provisional diagnosis of coxa vara in two patients, cubitus varus in one patient and Blount’s disease in one patient. A total of four patients had undergone vitamin D and calcium supplementation as treatment for presumed rickets.
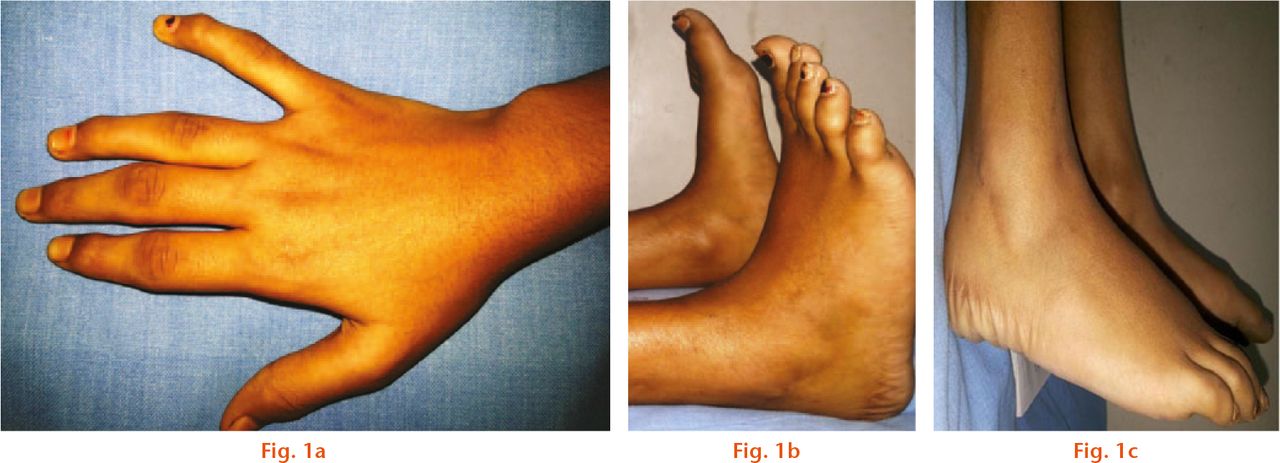
Fig.
Clinical photographs of a 13-year-old girl, which show that a) unique to PPD, the limitation of ankle movement is mainly in plantar flexion; b) classical epiphyseal swelling of the interphalangeal joints with stiffness but without pain is seen in the same patient; c) the same patient shows an enlarged posterior process of the talus which appears early and fuses before ten years of age.
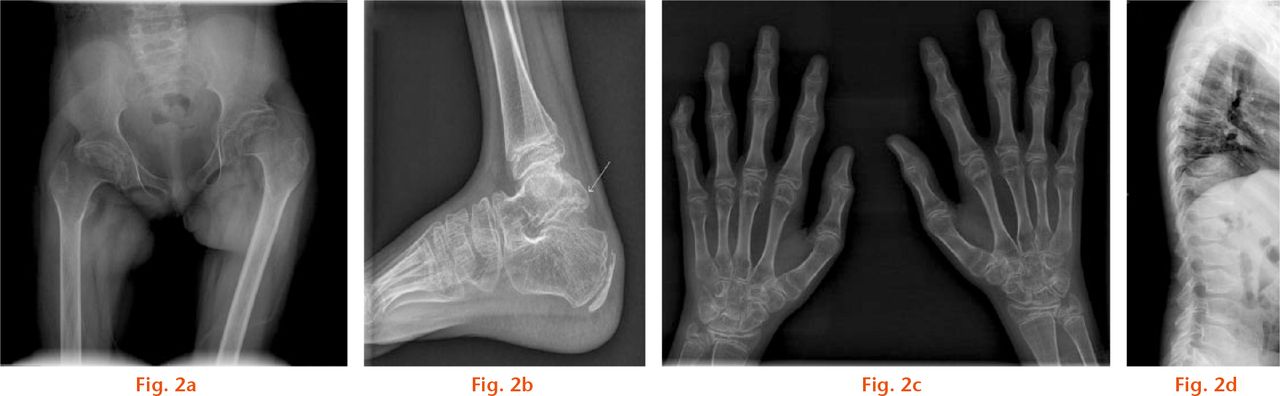
Fig.
Radiographs showing a) the pelvis of an 11-year-old girl showing lateralisation of the femoral head on the left side. There is altered growth on the capital femoral physis and flexion deformity. The hip protrusio is not seen, which distinguishes PPD from juvenile rheumatoid arthritis; b) the ankle of a nine-year-old boy shows near fusion of the enlarged posterior process of talus. Normal fusion of posterior process of the talus in boys usually occurs after 11 years; c) hand of an 11-year-old girl showing metaphyseal widening and epiphyseal shortening most prominent in the proximal interphalangeal joints and flexion deformity of the distal interphalangeal joint of the 5th digit, with an enlarged soft-tissue shadow with loss of joint space. The ulnar styloid process is hooked like a parrot’s beak in this child. The distal radial physis is flattened and the wrist joint is irregular; d) spine of 12-year-old girl showing platyspondyly with inferior and central beaking. There is a grade I L5-S1 spondylolisthesis. The lumbar vertebrae show inferior, and thoracic vertebrae show central, beaking.
Initial screening was done with CSGE for exon 5 and subsequently for the remaining exons. The mutated DNA sample formed a heteroduplex with the normal DNA and showed an abnormal banding pattern (Fig. 3). All 15 patients showed abnormal banding patterns, six in the second exon, two in the second and fourth exon, five in the fifth exon, and one each in the third and fourth exons. Mutations in the exons were confirmed by direct sequencing using Sanger’s method.
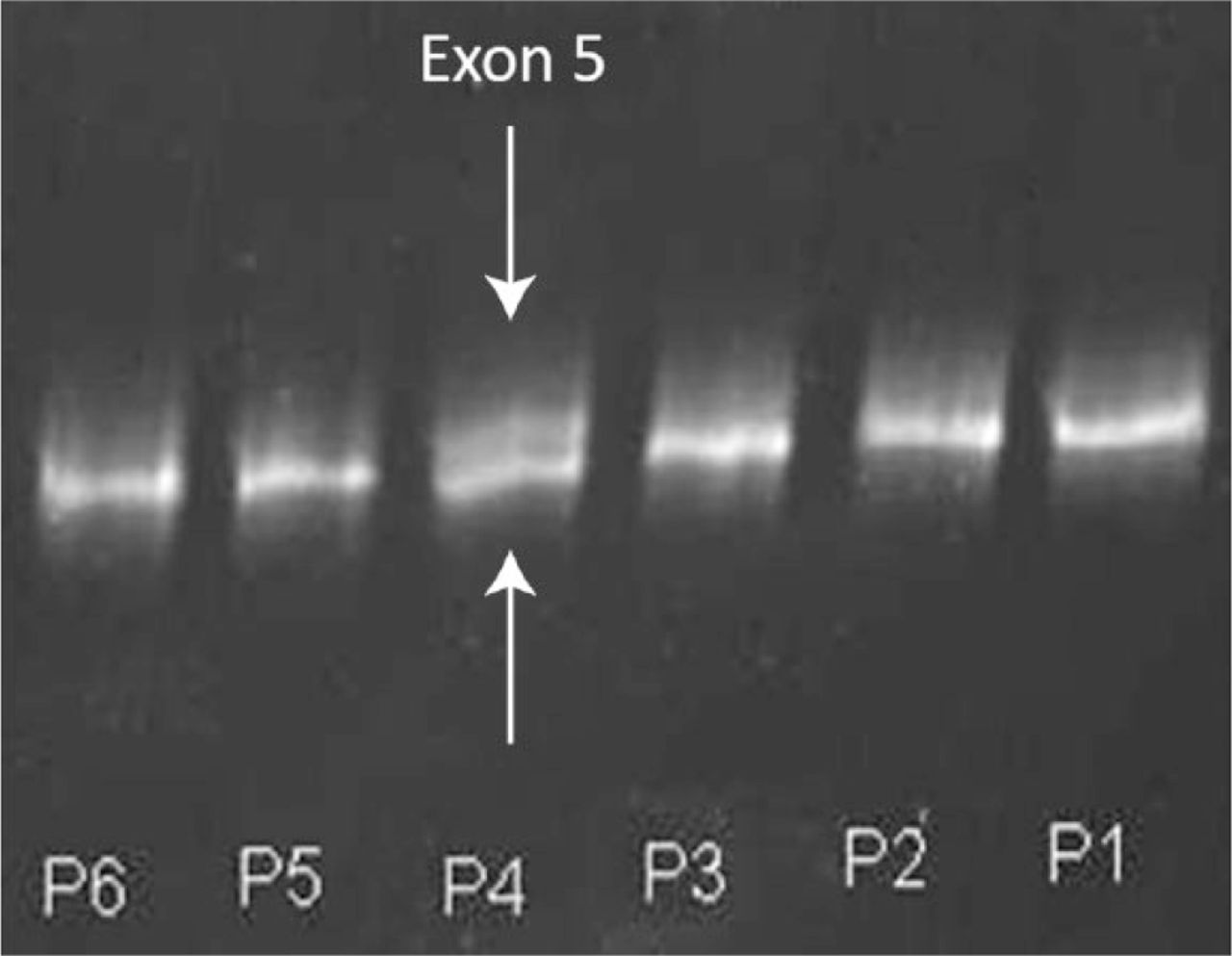
Fig. 3
Conformation-sensitive gel electrophoresis for six patients. The fourth patient (P4) shows a heteroduplex band indicated by the white arrow above the normal band of exon 5 (white arrow), indicating a mutation in the fifth exon of the WISP3 gene.
Sequence analysis confirmed the presence of a mutation in all 15 patients with suspected PPD (Fig. 4). In all, five types of point mutations were detected, including three missense, two nonsense and one deletion mutation (Table I). A total of five patients had the nonsense mutation c.156C>A (p.C52*) in exon 2. Two of these were heterozygous, with further analysis confirming the presence of another heterozygous mutation in c.677G>T (p.G226V) in exon 4. A c.233G>A (p.C78Y) missense mutation was observed in three patients and c.1010G>A (p.C337Y) mutation was identified in five patients. A nonsense mutation c.348C>A (p.Y116*) was detected in one patient. There was one mutation with a 5-nucleotide deletion in exon 4 in one patient. This novel mutation occurring at c.593_597delATAGA (p.Y198*) has not previously been described (Fig. 5). We confirmed that this novel mutation is a disease-causing mutation as it results in pre-termination of WISP3 protein synthesis (confirmed using Genetool Lite 1.0, Biotools Inc., Edmonton, Canada).
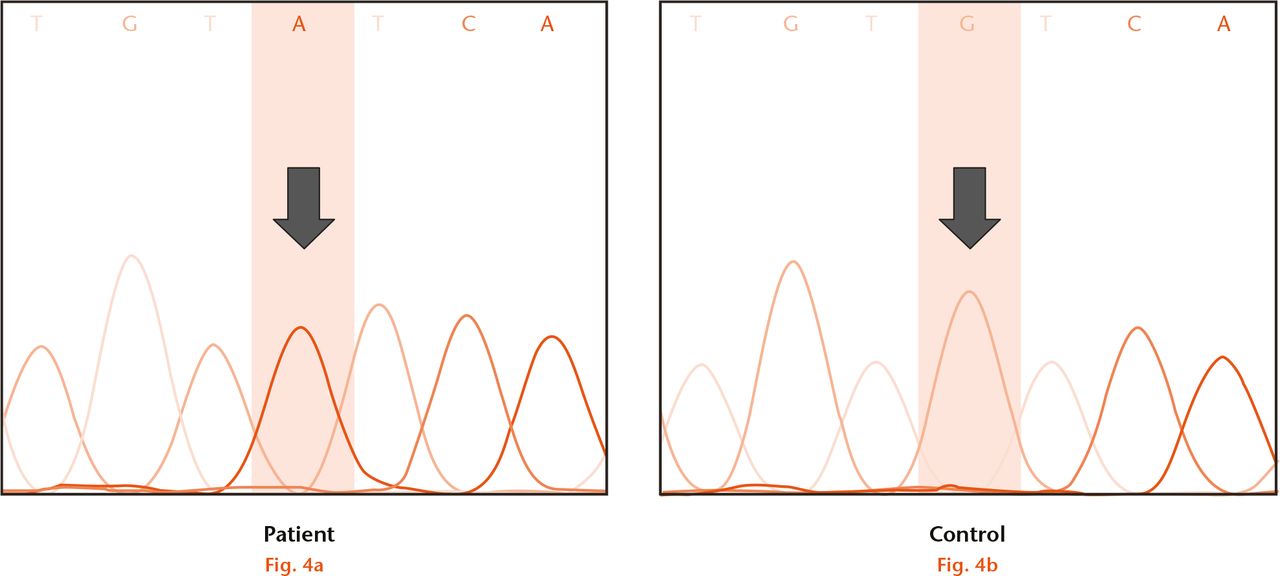
Fig.
Direct sequencing of fifth exon of the WISP3 gene in the fourth patient; (a) showed the presence of homozygous missense mutation c.1010G>A (p.C337Y) when compared with (b) control.
Table I.
Summary of all mutation in progressive pseudorheumatoid dysplasia patients
| Patient | Age (yrs) | Gender | Mutation in exon | Nucleotide change | Amino Acid change | Type of mutation | Allele state |
|---|---|---|---|---|---|---|---|
| 1 | 10 | M | 2 | c.156C>A | C52* | Nonsense | Homozygous |
| 2 | 13 | F | 2 | c.156C>A | C52* | Nonsense | Homozygous |
| 3 | 9 | F | 2 | c.156C>A | C52* | Nonsense | Homozygous |
| 4 | 10 | F | 5 | c.1010G>A | C337Y | Missense | Homozygous |
| 5 | 11 | M | 2 | c.233G>A | C78Y | Missense | Homozygous |
| 6 | 10 | M | 3 | c.348C>A | Y116* | Nonsense | Homozygous |
| 7 | 12 | F | 5 | c.1010G>A | C337Y | Missense | Homozygous |
| 8 | 14 | M | 5 | c.1010G>A | C337Y | Missense | Homozygous |
| 9 | 10 | M | 5 | c.1010G>A | C337Y | Missense | Homozygous |
| 10 | 9 | M | 5 | c.1010G>A | C337Y | Missense | Homozygous |
| 11 | 10 | F | 2 | c.156C>A | C52* | Nonsense | Compound Heterozygous |
| 4 | c.677G>T | p.G226V | Missense | ||||
| 12 | 13 | M | 2 | c.156C>A | C52* | Nonsense | Compound Heterozygous |
| 4 | c.677G>T | p.G226V | Missense | ||||
| 13 | 12 | F | 2 | c.233G>A | C78Y | Missense | Homozygous |
| 14 | 9 | F | 2 | c.233G>A | C78Y | Missense | Homozygous |
| 15 | 13 | F | 4 | c.593_597delATAGA | p.Y198* | Deletion | Homozygous |
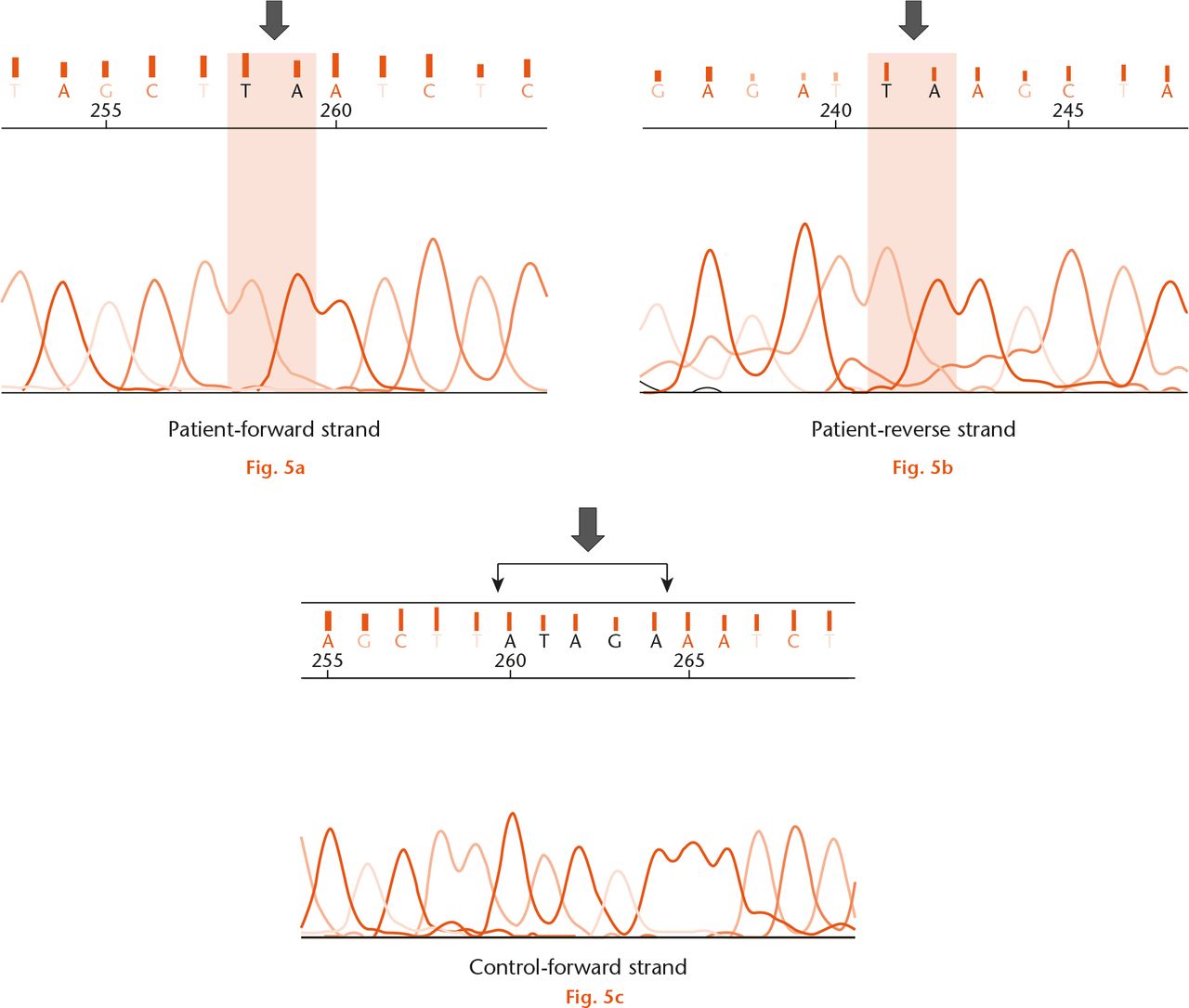
Fig.
Direct sequencing of fourth exon of the WISP 3 gene in the fifteenth patient shows (a) the absence of ATAGA sequence (black arrow) in the forward strand, indicating a deletion mutation c. 593_597delATAGA (p.Y198*); b) confirmation of this mutation was obtained by sequencing the reverse strand and; c) normal ATAGA sequence was seen in the control sample (black arrow).
Discussion
WISP3 encodes an intercellular signaling protein belonging to the CCN protein family and has a striking tetramodular structural organisation very similar to connective tissue growth factor.21 CCN proteins are involved in the regulation of mitosis, adhesion, apoptosis, extracellular matrix production, growth arrest and migration in multiple cell types.9
WISP3 is mutated in progressive pseudorheumatoid dysplasia and may have effects on cartilage homeostasis.8WISP3, also known as CCN6, is highly expressed in end-stage osteoarthritic cartilage, suggesting a role for this growth factor in cartilage homeostasis.22 The WISP3 gene is located on chromosome 6q22 and encodes a 354 amino acid protein.11WISP3 protein is highly expressed in skeletal cells and tissues and is important in regulating the expression of cartilage-specific molecules that sustain chondrocyte growth and cartilage integrity.1 Studies on the molecular mechanism of WISP3 suggest that, following secretion by chondrocytes, the protein can function via autocrine or paracrine modes to regulate collagen II and aggrecan expression and promote superoxide dismutase expression and activity in chondrocytes, thus enhancing its protective effect.23
Currently, 60 different mutations have been reported in the WISP3 gene in PPD patients around the world. Sun et al11 reported mutation c.156C>A(p.C52*) as a hot-spot mutation which was frequent in different ethnic groups from Turkey, Lebanon, Syria, Italy, and France.1,13,15 This mutation was identified in five of our patients. Dalal et al14 have reported this mutation in four of their patients, suggesting that this mutation is common to the Indian population. We found five distinct point mutations in our patients. All five types of point mutation detected in our study have been previously reported (Table II). Of these, the mutations c.233G>A (p.C78Y), c.348C>A (p.Y116*) and c.1010G>A (p.C337Y) were recently found in the Indian population.14 One of our female patients who presented at the age of 13 years with an inability to walk and rapidly progressing hip symptoms had a deletion in exon 4 at c.593_597delATAGA (p.Y198*). This is a deletion that has not been previously reported, and adds to the nine deletions, varying from one to seven nucleotides, that have been described in WISP3 thus far. The child with this novel mutation had a five nucleotide deletion resulting in a frame shift, thus leading to protein truncation or nonsense mediated decay.
Recently, eight novel mutations have been reported from the Indian population. A total of three of these, in locations p.C78Y, p.Y116* and p.C337Y, were present in our study.14 A total of five out of 15 patients in our study demonstrated c.1010G>A (p.C337Y). The latter appears to be a hot-spot mutation in the Indian population, having been reported in ten out of 25 families in the other Indian series.14
The clinical features of PPD mimic those of metabolic disorders such as rickets and those of juvenile rheumatoid arthritis.3 Even in the absence of inflammatory parameters, the condition is often missed unless the level of awareness is high. In our series, a mean of six and a half years elapsed between the onset of symptoms and diagnosis. A few radiological features such as joint space narrowing with flattened epiphyses and widened metaphyses, early appearance and fusion of the os trigonum and platyspondyly should alert the clinician to the diagnosis of this difficult disorder. Early and accurate molecular diagnosis of PPD requires screening of only five exons.11 Hence it is a feasible proposition and should be carried out in children with early onset deforming arthritis who lack clinical and laboratory evidence of a truly inflammatory process.
The common mutations in our series are distinct from those reported elsewhere but match those recently described in the two previous studies from this region.14,18 Further studies are required to comprehensively identify the mutation profile of PPD patients from this country.
Supplementary material
Tables showing WISP3 primers and all reported WISP3 mutations can be found alongside this paper at http://www.bjr.boneandjoint.org.uk/
Funding Statement
V. Madhuri reports receipt of a grant received from the Christian Medical College to undertake this study.
ICMJE conflict of interest
None declared.
References
1 Delague V , ChoueryE, CorbaniS, et al.. Molecular study of WISP3 in nine families originating from the Middle-East and presenting with progressive pseudorheumatoid dysplasia: identification of two novel mutations, and description of a founder effect. Am J Med Genet A2005;138A:118-126.CrossrefPubMed Google Scholar
2 Marik I , MarikovaO, ZemkovaD, KuklikM, KozlowskiK. Dominantly inherited progressive pseudorheumatoid dysplasia with hypoplastic toes. Skeletal Radiol2004;33:157-164.CrossrefPubMed Google Scholar
3 Rezai-Delui H , MamooriG, Sadri-MahvelatiE, NooriNM. Progressive pseudorheumatoid chondrodysplasia: a report of nine cases in three families. Skeletal Radiol1994;23:411-419.CrossrefPubMed Google Scholar
4 Mampaey S , VanhoenackerF, BovenK, Van HulW, De SchepperA. Progressive pseudorheumatoid dysplasia. Eur Radiol2000;10:1832-1835.CrossrefPubMed Google Scholar
5 Cogulu O , OzkinayF, OzkinayC, et al.. Progressive pseudorheumatoid arthropathy of childhood. Indian J Pediatr1999;66:455-460.CrossrefPubMed Google Scholar
6 Adak B , TekeoğluI, SakaryaME, UğrasşS. Progressive pseudorheumatoid chondrodysplasia: a hereditary disorder simulating rheumatoid arthritis. Clin Rheumatol1998;17:343-345.CrossrefPubMed Google Scholar
7 Ehl S , UhlM, BernerR, et al.. Clinical, radiographic, and genetic diagnosis of progressive pseudorheumatoid dysplasia in a patient with severe polyarthropathy. Rheumatol Int2004;24:53-56.CrossrefPubMed Google Scholar
8 Kutz WE , GongY, WarmanML. WISP3, the gene responsible for the human skeletal disease progressive pseudorheumatoid dysplasia, is not essential for skeletal function in mice. Mol Cell Biol2005;25:414-421. Google Scholar
9 Brigstock DR . The CCN family: a new stimulus package. J Endocrinol2003;178:169-175.CrossrefPubMed Google Scholar
10 Lau LF , LamSC-T. The CCN family of angiogenic regulators: the integrin connection. Exp Cell Res1999;248:44-57.CrossrefPubMed Google Scholar
11 Sun J , XiaW, HeS, et al.. Novel and recurrent mutations of WISP3 in two Chinese families with progressive pseudorheumatoid dysplasia. PLoS One2012;7:e38643.CrossrefPubMed Google Scholar
12 Sen M , ChengYH, GoldringMB, LotzMK, CarsonDA. WISP3-dependent regulation of type II collagen and aggrecan production in chondrocytes. Arthritis Rheum2004;50:488-497.CrossrefPubMed Google Scholar
13 Hurvitz JR , SuwairiWM, Van HulW, et al.. Mutations in the CCN gene family member WISP3 cause progressive pseudorheumatoid dysplasia. Nat Genet1999;23:94-98.CrossrefPubMed Google Scholar
14 Dalal A , BhavaniG SL, TogarratiPP, et al.. Analysis of the WISP3 gene in Indian families with progressive pseudorheumatoid dysplasia. Am J Med Genet A2012;158A:2820-2828.CrossrefPubMed Google Scholar
15 Temiz F , OzbekMN, KotanD, et al.. A homozygous recurring mutation in WISP3 causing progressive pseudorheumatoid arthropathy. J Pediatr Endocrinol Metab2011;24:105-108.CrossrefPubMed Google Scholar
16 Ye J , ZhangHW, WangT, et al.. Clinical diagnosis and WISP3 gene mutation analysis for progressive pseudorheumatoid dysplasia. Zhonghua Er Ke Za Zhi2010;48:194-198. (In Chinese). Google Scholar
17 Yue H , ZhangZ-L, HeJ-W. Identification of novel mutations in WISP3 gene in two unrelated Chinese families with progressive pseudorheumatoid dysplasia. Bone2009;44:547-554.CrossrefPubMed Google Scholar
18 Bhavani GS , ShahH, DalalAB, et al.. Novel and recurrent mutations in WISP3 and an atypical phenotype. Am J Med Genet A2015;167A:2481-2484.CrossrefPubMed Google Scholar
19 Garcia Segarra N , MittazL, Campos-XavierAB, et al.. The diagnostic challenge of progressive pseudorheumatoid dysplasia (PPRD): a review of clinical features, radiographic features, and WISP3 mutations in 63 affected individuals. Am J Med Genet C Semin Med Genet2012;160C:217-229.CrossrefPubMed Google Scholar
20 No authors listed. Primer-BLAST. http://www.ncbi.nlm.nih.gov/tools/primer-blast/ (date last accessed 28 June 2016).[[bibmisc]]CrossrefPubMed Google Scholar
21 Perbal B . 7th international workshop on the CCN family of genes: nice to be in nice. J Cell Commun Signal2013;7:165-167.CrossrefPubMed Google Scholar
22 Baker N , SharpeP, CulleyK, et al.. Dual regulation of metalloproteinase expression in chondrocytes by Wnt-1-inducible signaling pathway protein 3/CCN6. Arthritis Rheum2012;64:2289-2299.CrossrefPubMed Google Scholar
23 Davis L , ChenY, SenM. WISP-3 functions as a ligand and promotes superoxide dismutase activity. Biochem Biophys Res Commun2006;342:259-265.CrossrefPubMed Google Scholar
