




Abstract
This article provides an overview of the role of genomics in sarcomas and describes how new methods of analysis and comparative screening have provided the potential to progress understanding and treatment of sarcoma. This article reviews genomic techniques, the evolution of the use of genomics in cancer, the current state of genomic analysis, and also provides an overview of the medical, social and economic implications of recent genomic advances.
There have been significant recent advances in our understanding of the molecular nature of cancer, driven by progress in all aspects of genomics. Genomics is a convergence of many disciplines including genetics, molecular biology, biochemistry, statistics and computer sciences. It is now economically and scientifically feasible to analyse the whole human genome of affected cancer cells for comparison with the host germline (normal cells) in cancer patients. This sequencing comparison, available for a few thousand Australian dollars, allows the individual genetic changes associated with an individual's cancer to be identified. This information will enable improved treatment immediately in some patients by reference to current libraries, but carries the potential for personalised genetic diagnostics and tailored therapies. How much and how quickly our patients benefit from this conceptual potential will depend on how we adapt as a profession and a society to the challenges this programme brings.
The human genome is comprised of over three billion base pairs with approximately 20 000 genes, and the storage of this volume of information requires super computers. If widespread comparative analysis becomes commonplace, we are likely to discover novel mutations, particularly unstable ones that we will struggle to understand. However, even with our current knowledge, we can improve treatment in some patients and inform other patients at risk of developing certain cancers and the risk to their families. The conflict of personal privacy versus open disclosure to life insurance and other companies has brought into sharp focus the ethical difficulties that can disadvantage the patient unless genetic bias and discrimination is protected against in law. While genomics is relevant to many aspects of medicine, it has particular potential for sarcoma patients, and in this article we will look at traditional and developing techniques in genomics and how they relate to our sarcoma patients.
Recent history of genomics
The history of genomics is a convergence of the history of genetics and molecular biology. Gregor Mendel was the first to describe a unit of heredity; it was another 40 years before his work became widely cited1 and it took until the 1900s for Mendel's principles to become accepted. The chromosomal theory of inheritance was proposed independently in 1902 by Sutton and Boveri2,3 as the concept of hereditary information gained traction. Our understanding deepened rapidly and in 1905 the concept of linkage was demonstrated by Bateson and Punnett.4 Linkage is the close physical proximity of two or more genes on a chromosome which results in them being more likely to be inherited together. The DNA helix structure was first described by James Watson and Francis Crick in 1953,5 a discovery that gave birth to the modern age in genetics. The discovery of the DNA double helix has opened the door to understanding the mechanism for the flow of biological information through the carefully orchestrated ribosomal copying mechanism based on the specific pairing of nucleotide subunits.
The first karyotype depicting the correct number and appearance of chromosomes in the nucleus of a human cell was published just three years later in 1956 and identified 46 chromosomes.6 By 1972 the collective efforts of the scientific community led to the first gene sequencing from the bacteriophage, MS2.7 The first entire complete genome was sequenced in Cambridge by the Sanger group, whose early work focused on sequencing the genomes of a virus and mitochondrion in the early 1970s.8 In 1977 the chain-termination method of DNA sequencing was also published, marking a move towards the modern age of DNA sequencing.9 The use of automated DNA sequencing and polymerase chain reaction (PCR) was developed in the early 1980s and allowed DNA amplification from extremely small amounts of material,10,11 greatly increasing the potential applications of this early technology. In 1990 the technologies had advanced sufficiently to allow the Human Genome Project to be launched. The National Institute of Health (NIH) Human Genome Project was an international, collaborative research programme with the aim of complete mapping and understanding of all the genes present in humans. The product of the Human Genome Project was a resource of information about the structure, organisation and function of human genes. The International Human Genome Sequencing Consortium published the first draft of the human genome in 2001,12 with the sequence approximately 90% complete. In 2001 it was estimated that the genome contained between 35 000 and 40 000 genes. The full sequence was completed in April 2003. The stage had been set, this complete sequence (and the many more that have followed) has allowed for the birth of genomics and the understanding of heritable and other diseases at protein and genetic level.
With the completion of the Human Genome Project our understanding of the genetics of common diseases is increasing and many common genetic changes that predispose disease have been identified. It is now possible for individuals to get their own genomes sequenced commercially and have their risk of cancer and other diseases estimated based on current understanding of risk factors. Genetic testing is becoming increasingly important in cancer and with the decreased cost of genomic analysis that can identify individuals at higher risk and increase the effectiveness of targeted screening and preventative strategies.
Techniques used in genomic analysis
Cytogenetics
Cytogenetics is the branch of genetics concerned with the study of chromosomes and chromosomal abnormalities. In the 1970s, the evolution of staining techniques able to produce chromosome bands and to detect genetic deletions, duplications and other abnormalities, gave birth to cytogenetics. Giemsa banding (G-banding) is the most widely used banding process where the Giemsa stain is applied after chromosomal proteins are partially digested by trypsin. G-banding can be used to study the entire genome of individual cells for 'top down' chromosomal level changes. High resolution banding involves staining of chromosomes during prophase or early metaphase, however, cytogenetic analysis is not considered to be high resolution. Each band represents approximately 5 to 10 x 106 base pairs and only major structural changes can be demonstrated with this technique.13 Chromosome banding analysis is typically used for karyotypic abnormalities such as differences in absolute size of chromosomes, position of centromeres, relative size of chromosomes, basic number of chromosomes, number and position of satellites, and the degree and distribution of heterochromatic regions. An example of the use of cytogenetics in detecting chromosomal abnormalities is Ewing's sarcoma. A majority of Ewing's sarcoma cases are the result of a translocation t(11:22)(q24:q12)14 resulting in a fusion of the genes EWS and FLI1.15
Fluorescent in situ hybridisation
Fluorescent in situ hybridisation (FISH) is a molecular cytogenetic technique used to provide cellular localisation. Chromosome-specific probes are labelled with fluorescent dyes and exposed to denatured chromosome material during metaphase, prophase or interphase. As the probe is labelled with fluorescent dye, the chromosomes can be viewed under a fluorescent microscope. Probes can be centromere-specific, locus-specific, or 'paint' probes, where whole chromosomes are labelled. FISH provides better resolution than high resolution banding techniques, able to detect deletions as small as one million base pairs. Novel FISH approaches such as multi-colour FISH, spectral karyotyping, combined binary ratio FISH, metaphase-based comparative genomic hybridisation (CGH), array-based CGH and SNP arrays are genome-wide applications. FISH-mediated detection of MDM2amplification is a valuable diagnostic aid for atypical lipomatous tumour/well-differentiated liposarcoma and de-differentiated liposarcoma 16
Analysis of gene expression
Gene expression is the process by which information from a gene is translated into a functional product. Regulation of genetic expression can occur at this stage prior to transcription as post-transcriptional regulation. Analysis of gene expression has become one of the most widely used strategies for discovering and understanding mechanisms underlying cancer.17 Analysis of mRNA and proteins can be used to compare patterns of gene expression between cells or tissues, for example, germline and cancer cells.
Over the past four decades, several methods have been developed to allow for comparative studies of gene expression, often between germline and cancer cells. The first two-dimensional gel electrophoresis was developed by O'Farrell in 1975 and allowed the visualisation of protein expression.18 This method was successful in discovering the p53 tumour-suppressor protein.19 Analysis of gene expression evolved to analysing mRNA expression using complementary DNA. A much more advanced technique whereby comparison of hybridisation patterns allowed identification of genes that were uniquely expressed in one sample but not the other.20
Differential display was developed in the 1990s, which integrated polymerase chain reaction (PCR) and DNA sequencing by gel electrophoresis.21 Several oncogene targets have been identified by differential display including RAS, v-REL and ERBB,22-25 as well as several target genes of the p53 tumour suppressor.
Expressed sequence tags (EST) focused on sequencing expressed genes rather than the whole genome.26 EST played a major role in gene discovery for the NIH Cancer Genome Anatomy Program which provides cDNA clones for functional studies of genes. Serial analysis of gene expression (SAGE), developed in the 1990s,27 is an open system based gene discovery tool. The NIH Cancer Genome Project has a comprehensive SAGE database for normal and cancer cell lines and tissues. Massively parallel signature sequencing (MPSS) combines non-gel-based signature sequencing with in vitro cloning of millions of templates on separate 5 μ diameter microbeads.28
Automated microarray analysis
Perhaps the biggest leap forward in genetic analysis is the development of DNA microarrays which may be used to perform an automated measure of the expression levels of large numbers of genes simultaneously or to genotype multiple regions. Advances in microarray technology enable massive parallel mining of biological data, with biological chips providing hybridisation-based expression monitoring, polymorphism detection and genotyping on a genomic scale. The application of microarray technology to assess mRNA on a genome-wide scale has resulted in large data sets in themselves posing unique problems. The vast quantity of data required new tools for analysing results, such as automated microarray analysis software (AMDA),29 automated microarray image analysis (AMIA)30 and automated gene ontology analysis of expression profiles (GOAL).31
Microarray methods were initially developed to study differential gene expression using complex populations of RNA,32 but like all technologies have developed, and advances now permit analysis of copy number variants and gene amplifications,33 as well as being able to quantify gene expression at the protein level.34 Microarray technology has been widely used to investigate tumour classification, cancer progression, chemotherapy and other drug resistance and sensitivity, and identification of tumour-specific molecular markers.35 Histologically indistinguishable tumours often show differences in clinical behaviour such as response to treatment. Sub-classification of these tumours based on their molecular profiles may help explain these differences and ultimately identify novel therapeutic targets and offer the potential for targeted therapies based on the molecular 'signature' of individual tumours.
Microarrays can be used to study the molecular pathways implicated in mechanisms that determine anticancer drug resistance, and to this end the majority of array studies have been carried out using cancer cell lines that are rendered resistant to commonly used anticancer drugs.35 One of the most useful applications of microarray technology is in the identification of genes that exhibit differential expression between healthy tissues or cell lines and their tumour counterparts. Successful identification of differential gene expression is suggestive of a potential molecular marker or even a potential therapeutic intervention point.35 Microarray analysis may provide invaluable information on disease pathology, progression, resistance to treatment, and response to cellular microenvironments that may lead to improved early diagnosis and innovative therapeutic approaches for cancer. Genome-wide complementary DNA microarray was recently used to characterise the gene expression profile of pigmented villonodular synovitis (PVNS) in comparison with osteoarthritis and rheumatoid arthritis.36 Genes that were differentially expressed in PVNS were involved in apoptosis regulation, matrix degradation and inflammation,36 all of which are now potential sites for pharmacological intervention.
Bioinformatics
Bioinformatics is the science of storing, retrieving, organising and analysing biological data, perhaps the most important part of genomics with the value of any data being determined by the strength of the interpretation. Genetic applications of bioinformatics include sequencing and annotating genomes and mutations. Sequence information can serve to determine which genes encode proteins, RNA, regulatory sequences, structural motifs and repetitive sequences. The comparison of these features is often the starting point in determining gene similarities or relations. Annotation is the process of marking genes and other biological features found in genetic material and is aided greatly by the use of computer programmes such as basic local alignment search tool (BLAST). These software tools are used to search known sequences from more than 250 000 organisms.37 BLAST provides sequence similarity searches of GenBank and other sequence databases. In cancer research, bioinformatics is used to store and organise information regarding the increasing number of mutations and rearrangements that have been characterised and comparing sequencing results with human genome sequences to determine germline polymorphisms. Massive sequencing efforts have been used to identify new point mutations requiring bioinformaticians to develop systems that manage the volume of data produced, and then to develop new algorithms and software to compare the results. A prime example of cancer bioinformatics in action is The Cancer Genome Atlas (TCGA) data portal (National Cancer Institute 2013)38(Fig. 1). The portal provides a platform for researchers to search, download, and analyse data sets generated by TCGA. The use of high throughput technologies and bioinformatics is important for heterogeneous groups of tumours, such as sarcomas, to distinguish histologically similar but molecularly different types.
Fig. 1
Image of the Cancer Genome Atlas Portal.
Types of mutation found in cancer
A cell becomes cancerous only after changes occur in a number of genes that are involved in the regulation of its cell cycle. Proto-oncogenes and tumour-suppressor genes are the two most common types of gene involved in regulating the cell cycle. Proto-oncogenes are responsible for promoting the controlled growth and division of cells. When proto-oncogenes are mutated they result in oncogenes that produce an abnormal protein stimulating uncontrolled cell growth. Tumour-suppressor genes produce proteins that regulate the cell cycle and are involved in helping to prevent uncontrolled cell growth and division. When a tumour suppressor-gene is mutated this can result in altered or absent function including loss of an important regulator of mitosis, and open the door for tumour formation or growth. In this complex interaction the function of DNA repair genes is also critical. These genes work to correct errors that occur during DNA polymerisation and cell division. Loss of function of DNA repair genes has obvious consequences. Oncogenes, tumour-suppressor genes and DNA repair genes can be altered or mutated by chromosome rearrangements, gene duplication or mutation. Additionally, the function of regulatory genes can be altered by changes in promoter regions regulating the expression of a particular gene or group of genes. A chromosome rearrangement involving a tumour-suppressor gene or proto-oncogene can contribute to the transformation of a normal cell into a cancerous cell. There are several types of chromosome rearrangements; deletions, duplications, inversions and translocations.
A deletion is the loss of part of DNA sequences and the effects of a deletion depend on the location, function of and size of the deleted sequence. Deletions not involving three (or multiples thereof) nucleotides can also result in the 'frame-shift' effect where all of the downstream genetic information is altered as nucleotides function in triplets. Duplications occur when an extra copy of a chromosomal region is produced and again the location of the duplication has a critical effect on the functional consequences. In cancer, deletions and copy number increases contribute to alterations in the expression of tumour-suppressor genes and oncogenes, respectively.
Inversions and translocations do not involve the loss or gain of genetic material, unlike deletions and duplications. If inversions or translocations occur within genes, they may affect gene function or a novel gene fusion may be produced. Inversions are rearrangements where a segment of DNA is broken, flipped and rejoined. Translocations involve the exchange of chromosomal material between two or more non-homologous chromosomes. Approximately one third of all sarcomas exhibit a non-random chromosomal translocation. A fusion gene results in the expression of multiple genes in response to the activation of a single promoter region. In soft-tissue tumours, chromosome translocations commonly result in highly specific, novel chimeric genes.13 An example is the reciprocal translocation found in synovial sarcoma, t(x;18)(p11;q11) and the presence of a SYT-SSX fusion gene.39
Stable versus unstable
One of the hallmarks of cancer cells is genetic instability. Instability can be at the single nucleotide level resulting in a point mutation, or at a chromosome level such as translocations, deletions, amplifications or aneuploidy.40 DNA is continuously undergoing damage, repair and resynthesis. It is a homeostatic equilibrium where DNA damage is counterbalanced by DNA repair. In normal cells DNA damage is repaired without mistakes. In tumour cells this equilibrium is lost and over time multiple mutations accumulate. It is rare for cancer to develop with a single mutation; usually multiple genetic changes are required. The emerging concept is that genomes of cancer cells are unstable and this instability results in the accrual of a cascade of mutations that allows cancer cells to bypass regulatory processes that control cell location, division, expression, adaptation and death.40
Genomics and cancer
Genomic data, particularly gene expression signatures, can be used as clinical prognostic factors in cancer and other complex diseases. Genomics can be used to guide the use of currently available cancer drugs, develop new targeted therapeutics, and provide an opportunity to match the most effective drugs with the molecular characteristics of the individual patient. The most successful applications of genomic technology have been in the study of human cancer, in which gene expression patterns can be identified that provide phenotypic detail not previously obtained by traditional methods of analysis; profiles and patterns that identify new subclasses of tumours. A recent example includes the distinction between acute myeloid leukaemia and acute lymphoblastic leukaemia.41 Genomic techniques may also be useful in determining more targeted applications for existing cancer therapeutics, many of which are very effective for subsets of cancer patients, thereby further ensuring treatment becomes tailored to the individual.
Recent years have seen significant advances in our understanding of the molecular nature of cancer driven by advances in high throughput technologies and DNA sequencing technologies. Genomic changes can be used as molecular biomarkers to identify patients who may respond to a treatment. Many cancer drugs have not been linked to specific markers yet there is still a need for development of biomarkers to guide therapy and improve outcomes.
Evolution of genomic analysis in sarcoma
Sarcomas are uncommon mesenchymal malignancies that arise in bone, cartilage or connective tissue; they have extremely varied genetic origins, are clinically heterogeneous (Fig. 2). Many sarcomas arise de novo. Sarcomas can be genetically classified into two categories; those with near-diploid karyotypes and simple genetic alterations including translocations, and a second category of sarcoma with complex and unbalanced karyotypes.42
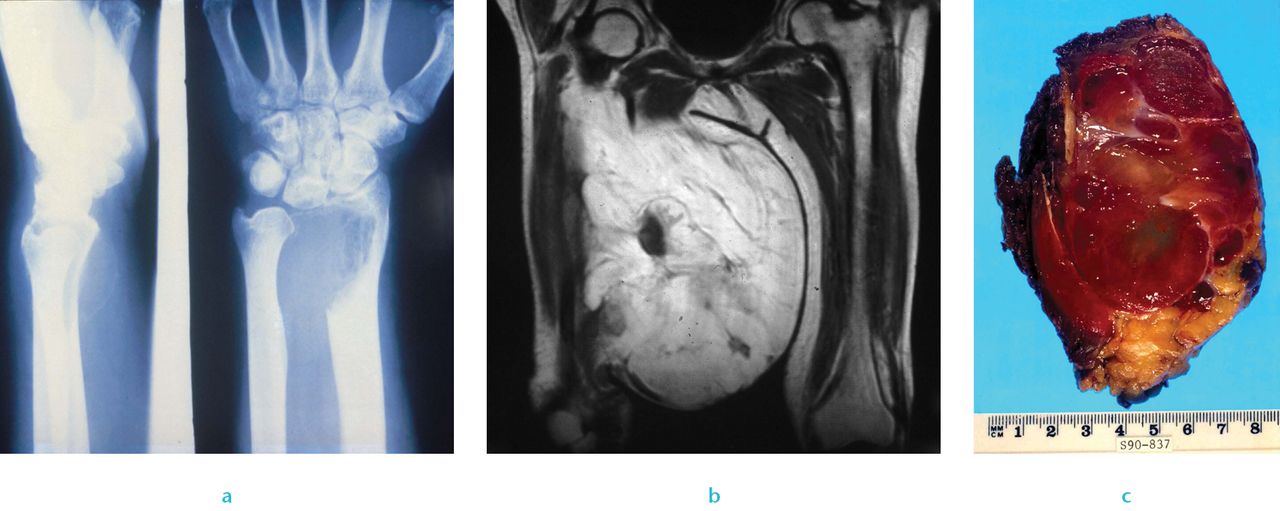
Fig. 2
Clinical sarcoma: a) Radiograph of a giant cell tumour of bone, b) imaging of myxoid liposarcoma and c) image of liposarcoma.
Those with complex and unbalanced karyotypes are typified by high levels of genomic instability, resulting in multiple genetic aberrations in the genome of a single tumour and heterogeneity of aberrations across tumours of a given type.42 Most simple genetic alterations seen in sarcomas are translocation-associated.42 Gene fusions resulting from these translocations typically encode chimeric transcription factors that cause transcriptional deregulation (loss of control of gene expression) of target genes. Less commonly they encode chimeric protein tyrosine kinases or autocrine growth factors.43 Karyotypically complex sarcomas can begin as a less aggressive form and then progress through discrete stages with increasing genomic complexity,42 for example, atypical lipoma or well-differentiated liposarcoma progress to dedifferentiated liposarcoma.44
There are three categories of mechanisms that drive sarcomagenesis; transcriptional dysregulation owing to aberrant fusion proteins that result from genomic arrangements, somatic mutations in key genes and signalling pathways, and DNA copy number abnormalities.42 Transcriptional target gene dysregulation in translocation sarcomas has been the focus of the application of genome-wide transcription factor location analysis to identify target genes of fusion proteins, as well as emerging evidence for abnormal nuclear reprogramming of mesenchymal stem cells in translocation-associated sarcomas.42
Another recent advance is the use of transcriptional targets of fusion proteins as therapeutic agents. An example of mutations in key genes and signalling pathways is gastrointestinal stromal tumour (GIST), characterised by oncogenic mutations in KIT, PDGRF α and BRAF.45-47The implication of KIT and PDGRF α has led to treatment of GIST with selective kinase inhibitors, an example of a direct therapy 'designed' from the ground up based on genomic study of GIST cells.
Systematic surveys of cancer genomes with integrated genomics have been used to identify targeted genetic alterations in cancer. Targets are expected to grow with the use of second-generation sequencing technologies. The Cancer Genomic Atlas (TCGA) is beginning a comprehensive genomic analysis of dedifferentiated liposarcoma, leiomyosarcoma and undifferentiated pleomorphic sarcoma.48
Current technology
Efficiency of whole human genome sequencing versus exome sequencing
Full genome sequencing provides information on all six billion base pairs in the genome, whereas exome sequencing selectively sequences only at the coding regions of the genome. Exons are the regions of DNA that are translated into proteins. The goal of exome sequencing is to sequence the coding region that has the potential to be clinically relevant due to functional changes in the sequence of proteins. Whole genome sequencing not only sequences coding regions but also regulatory elements.
Costs
The first human genome was sequenced in 2003 at a cost of approximately $40 million AUSD, requiring international collaboration and supercomputing power. The development of new technologies including massively parallel sequencing systems has reduced the cost of whole human genome sequencing studies to approximately $2000 to $3000 AUSD per individual and likely to reduce further to $1000 AUSD per individual in the next year. Whole exome sequencing is approximately $500 AUSD per individual, and has become so accessible that it is possible to privately commercially sequence an individual's genome (www.23andme.com) bringing this technology into the online community.
Ethics
There are many ethical issues associated with genetic research including discrimination, patient privacy, consent and the reporting of incidental findings. As DNA testing increasingly identifies differences in the DNA sequence of individuals and potentially the likelihood of an individual to develop or pass on a certain disease or condition; it becomes possible to discriminate based solely on genetic information. Discrimination against individuals based on their genetic information could arise in a wide range of situations; currently health risks would be relevant to insurance companies or employers. However, it is not difficult to imagine a situation where genetic pre-disposition for anti-social behavioural traits would be of interest to landlords and law enforcement agencies, or 'genetic IQ' used in selection for university or employment. To protect against these situations before they arise, the Genetic Information Nondiscrimination Act was passed into law in 2008 in the United States.49The act prohibits discrimination in the workplace, and by health insurance companies. The act prohibits group health plans and insurers from denying coverage or charging higher premiums based solely on genetic predisposition to developing a disease in the future. 50 In Australia insurance companies cannot charge higher premiums due to a person's genetic information, however, genetic information about a person or their family can affect a person's application for life insurance products, such as cover for death and income protection because these types of insurance are risk rated and genetic information can be taken into account in applications.
Due to the potential impact of genetic testing, patients should be adequately counselled about the specifics of testing. Before an individual agrees to participate in a clinical trial, research project or undergo a genetic test, he or she must be informed of the test's purpose, medical implications, alternatives, and possible risks and benefits. Patients should additionally be made aware of their privacy rights, including where their DNA will be stored and who will have access to their personal information. Australian guidelines for the conduct of ethical research can be found in the National Statement on Ethical Conduct in Human Research.51
Whole genome sequencing and exome sequencing increase the possibility of encountering variants associated with disease outside the original intent of testing. In the case where research may discover or generate information of potential importance to the future health of participants, or their blood relatives, researchers must prepare and follow an ethically defensible plan to disclose or withhold that information. Ethical issues that arise when segments of a patient's genome are interrogated include the risk of providing patients with incomplete or incorrect information, providing information for which patients are not prepared, exposing patients to unnecessary, harmful or ineffective treatment, and determining whether or not to report misattributed paternity, consanguinity or carrier status.
Gene libraries for genomics of drug sensitivity
There is evidence that a patient's cancer response to treatment can be influenced by the cancer genome. Single gene mutations are increasingly being adopted as clinical biomarkers for the optimal application of cancer therapeutics. There is a need to develop biomarkers to optimise drug development and clinical use. Biomarkers are being used in the decision-making process in discovery stages and in assessing the performance of drugs in clinical studies.52 Prognostic biomarkers are those that determine patient selection for treatment based on an estimation of the natural history of disease. Predictive biomarkers are single mutations that can be used to provide estimations of the probability of response to a particular treatment. An example of the use of drugs selectively to target the protein product of the BCR-ABL translocation in chronic myeloid leukaemia (CML) has revolutionised the treatment of this disease, with five-year survival rates of 90% in treated patients.53 The NCI60 cell line panel and associated drug screens pioneered the approach of using cancer cell lines to link drug sensitivity with genotype data.54,55 Cancer cell lines have subsequently been used to identify rare drug-sensitising genotypes, including mutant EGFR, BRAF and the EML4-ALK translocation, which are highly predictive of clinical responses.56-58
The Genomics of Drug Sensitivity in Cancer (GDSC) database59 is the largest public resource for information on drug sensitivity in cancer cells and molecular markers of drug response. GDSC currently contains drug sensitivity data for almost 75 000 experiments, describing responses to 138 anticancer drugs across almost 700 cancer cell lines. GDSC provides a unique resource incorporating large drug sensitivity and genomic data sets to facilitate the discovery of new therapeutic biomarkers for cancer therapies.60
There are currently few specific genetic lesions in sarcoma that are direct targets of any therapy. The exception among sarcomas is GIST, in which the KIT kinase inhibitor imatinib achieves a partial response or stable disease in approximately 80% of patients with advanced or metastatic GIST, often within days, with some patients now on therapy for ten years.61 Functional studies of sarcoma to find targets for therapy are limited by two factors. The first is that only limited numbers of human sarcoma cell lines exist, partly because of the rarity of certain diagnoses and the resulting scarcity of samples. Secondly for each of the subtypes with complex genomes, multiple cell lines are needed to represent the diversity of genetic alterations within that subtype. The challenge for sarcoma biologists, oncologists and surgeons is to continue to push basic science forward and clinical studies, which will allow the novel technologies of genomics, to be applied to the sarcoma population.
Several studies have used cancer cell lines to link pharmacological data with genomic information, and have also helped define therapeutic biomarkers.53,58,62 Cancer cell line drug sensitivity data are generated by the Cancer Genome Project at the Wellcome Trust Sanger Institute (WTSI) and the Center for Molecular Therapeutics at Massachusetts General Hospital.60 Other gene libraries such as Cancer Cell Line Encyclopedia63and Sanger Cancer Cell Line Project64 are aiming to characterise large numbers of human cancer cell lines genetically and screen these against a range of anticancer therapies to correlate drug sensitivity with genetic markers.
Conclusion
New efficient methods of whole human genome analysis and comparative screening provide unparalleled potential to progress our understanding and treatment of cancer. We have a responsibility to manage the science, ethics and access new treatments that this technology will provide.
1 Mendel G. Verhandlungen des Naturforschenden Vereines in Brünn. Vol. 4. Brünn: Im Verlage des Vereines; Versuche über Pflanzen-Hybriden, 1866: 3–47. Google Scholar
2 Sutton W . The chromosomes in heredity. Biological Bulletin1903;4:231–251. Google Scholar
3 Boveri T. Ergebnisse über die Konstitution der chromatischen Substanz des Zellkerns. Germany: Jena G. Fischer 1904. Google Scholar
4 Punnett RC. Mendelism. Second ed. Cambridge: Bowes and Bowes, 1907. Google Scholar
5 Watson JD , CrickFHC. A structure for deoxyribose nucleic acid. Nature1953;421:397–398.CrossrefPubMed Google Scholar
6 Tjio JH . The chromosome number of man. Am J Obstet Gynecol1978;130:723–724.CrossrefPubMed Google Scholar
7 Jou WM , HaegemanG, YsebaertM, FiersW. Nucleotide sequence of the gene coding for the bacteriophage MS2 coat protein. Nature1972;237:82–88.CrossrefPubMed Google Scholar
8 Sanger F , AirGM, BarrellBG, et al.Nucleotide sequence of bacteriophage [phi]X174 DNA. Nature1977;265:687–695. Google Scholar
9 Sanger F , NicklenS, CoulsonAR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci1977;74:5463–5467.PubMed Google Scholar
10 Saiki RK , GelfandDH, StoffelS, et al.Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science1988;239:487–491.CrossrefPubMed Google Scholar
11 Saiki RK , ScharfS, FaloonaF, et al.Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science1985;230:1350–1354.PubMed Google Scholar
12 Lander ES , LintonLM, BirrenB, et al.Initial sequencing and analysis of the human genome. Nature2001;409:860–921.CrossrefPubMed Google Scholar
13 Bridge JA , Cushman-VokounAM. Molecular diagnostics of soft tissue tumors. Arch Pathol Lab Med2011;135:588–601.CrossrefPubMed Google Scholar
14 Turc-Carel C, Aurias A, Mugneret F, et al. Chromosomes in Ewing's sarcoma. I. An evaluation of 85 cases and remarkable consistency of t(11;22)(q24;q12). Cancer Genet Cytogenet 1988;32:229-38. Google Scholar
15 Sorensen PH, Lessnick SL, Lopez-Terrada D, et al. A second Ewing's sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet 1994;6:146-51. Google Scholar
16 Kimura H , DobashiY, NojimaT, et al.Utility of fluorescence in situ hybridization to detect MDM2 amplification in liposarcomas and their morphological mimics. Int J Clin Exp Pathol2013;6:1306–1316.PubMed Google Scholar
17 Liang P , PardeeAB. Analysing differential gene expression in cancer. Nat Rev Cancer2003;3:869–876.CrossrefPubMed Google Scholar
18 O'Farrell PH . High resolution two-dimensional electrophoresis of proteins. J Biol Chem1975;250:4007–4021. Google Scholar
19 Matlashewski G , LambP, PimD, et al.Isolation and characterization of a human p53 cDNA clone: expression of the human p53 gene. EMBO J1984;3:3257–3262.CrossrefPubMed Google Scholar
20 Sargent TD . Isolation of differentially expressed genes. Methods Enzymol1987;152:423–432.CrossrefPubMed Google Scholar
21 Liang P , PardeeAB. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science1992;257:967–971.CrossrefPubMed Google Scholar
22 McCarthy SA , SamuelsML, PritchardCA, AbrahamJA, McMahonM. Rapid induction of heparin-binding epidermal growth factor/diphtheria toxin receptor expression by Raf and Ras oncogenes. Genes Devt1995;9:1953–1964.CrossrefPubMed Google Scholar
23 Zhang R , TanZ, LiangP. Identification of a novel ligand-receptor pair constitutively activated by Ras oncogenes. J Biol Chem2000;275:24436–24443.CrossrefPubMed Google Scholar
24 You M , KuPT, HrdlickováR, Bose HR Jr. ch-IAP1, a member of the inhibitor-of-apoptosis protein family, is a mediator of the antiapoptotic activity of the v-Rel oncoprotein. Mol Cell Biol1997;17:7328–7341.CrossrefPubMed Google Scholar
25 Park BW , O'RourkeDM, WangQ, et al.Induction of the tat-binding protein 1 gene accompanies the disabling of oncogenic erbB receptor tyrosine kinases. Proc Natl Acad Sci1999;96:6434–6438.CrossrefPubMed Google Scholar
26 Adams MD , DubnickM, KerlavageAR, et al.Sequence identification of 2,375 human brain genes. Nature1992;355:632–634.CrossrefPubMed Google Scholar
27 Velculescu VE , ZhangL, VogelsteinB, KinzlerKW. Serial analysis of gene expression. Science1995;270:484–487.CrossrefPubMed Google Scholar
28 Brenner S , JohnsonM, BridghamJ, et al.Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat Biotechnol2000;18:360–364.CrossrefPubMed Google Scholar
29 Pelizzola M , PavelkaN, FotiM, Ricciardi-CastagnoliP. AMDA: an R package for the automated microarray data analysis. BMC Bioinformatics2006;7:335–344.CrossrefPubMed Google Scholar
30 White AM , DalyDS, WillseAR, ProticM, ChandlerDP. Automated microarray image analysis toolbox for MATLAB. Bioinformatics2005;21:3578–3579.CrossrefPubMed Google Scholar
31 Volinia S , EvangelistiR, FranciosoF, et al.GOAL: automated Gene Ontology analysis of expression profiles. Nucleic Acids Res2004;32(Web Server issue):492–499.CrossrefPubMed Google Scholar
32 Lipshutz RJ , FodorSPA, GingerasTR, LockhartDJ. High density synthetic oligonucleotide arrays. Nat Genet1999;21:20–24.CrossrefPubMed Google Scholar
33 Pollack JR , PerouCM, AlizadehAA, et al.Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet1999;23: 41-6. :.CrossrefPubMed Google Scholar
34 Haab BB . Advances in protein microarray technology for protein expression and interaction profiling. Curr Opin Drug Discov Devel2001;4:116–123.PubMed Google Scholar
35 Macgregor PF , SquireJA. Application of microarrays to the analysis of gene expression in cancer. Clin Chem2002;48: 1170–7.:.PubMed Google Scholar
36 Finis K , SültmannH, RuschhauptM, et al.Analysis of pigmented villonodular synovitis with genome-wide complementary DNA microarray and tissue array technology reveals insight into potential novel therapeutic approaches. Arthritis Rheum 2006;54:1009–1019.CrossrefPubMed Google Scholar
37 Benson DA , Karsch-MizrachiI, ClarkK, et al.GenBank. Nucleic Acids Res2012;40(Database issue):48–53.CrossrefPubMed Google Scholar
38 No authors listed. The Cancer Genome Atlas Data Portal. https://tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp (date last accessed 4 November 2013). Google Scholar
39 dos Santos NR , de BruijnDRH, van KesselAG. Molecular mechanisms underlying human synovial sarcoma development. Genes Chromosomes Cancer2001;30:1–14.CrossrefPubMed Google Scholar
40 Loeb KR , LoebLA. Significance of multiple mutations in cancer. Carcinogenesis2000;21:379–385.CrossrefPubMed Google Scholar
41 Garman KS , NevinsJR, PottiA. Genomic strategies for personalized cancer therapy. Hum Mol Genet2007;16:226–232.CrossrefPubMed Google Scholar
42 Taylor BS , BarretinaJ, MakiRG, et al.Advances in sarcoma genomics and new therapeutic agents. Nat Rev Cancer2011;11:541–557. Google Scholar
43 Mertens F , AntonescuCR, HohenbergerP, et al.Translocation-related sarcomas. Sem Oncol2009;36:312–323.CrossrefPubMed Google Scholar
44 Horvai AE , DeVriesS, RoyR, O'DonnellRJ, WaldmanF. Similarity in genetic alterations between paired well-differentiated and dedifferentiated components of dedifferentiated liposarcoma. Mod Pathol2009;22:1477–1488.CrossrefPubMed Google Scholar
45 Agaram NP , WongGC, GuoT, et al.Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer2008;47:853–859.CrossrefPubMed Google Scholar
46 Heinrich MC , CorlessCL, DuensingA, et al.PDGFRA activating mutations in gastrointestinal stromal tumors. Science2003;299:708–710.CrossrefPubMed Google Scholar
47 Hirota S , IsozakiK, MoriyamaY, et al.Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science1998;279:577–580.CrossrefPubMed Google Scholar
48 No authors listed. The Cancer Genome Atlas Data Portal. https://tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp (date last accessed 7 October 2013). Google Scholar
49 No authors listed. Genetic information nondiscrimination act of 2008. http://www.govtrack.us/congress/bills/110/hr493/text (date last accessed 7 October 2013) . Google Scholar
50 No authors listed. Genetic information nondiscrimination act of 2007. http://www.genome.gov/pages/policyethics/geneticdiscrimination/saponhr493.pdf. (date last accessed 7 October 2013). Google Scholar
51 No authors listed. National Statement on Ethical Conduct in Human Research. http://www.ambulance.vic.gov.au/media/docs/e72_national_statement_130624-028bd263-b454-4088-a6d0-64eff4ce07d2-0.pdf (date last accessed 31 October 2013). Google Scholar
52 Alymani NA , SmithMD, WilliamsDJ, PettyRD. Predictive biomarkers for personalised anti-cancer drug use: discovery to clinical implementation. Eur J Cancer2010;46:869–879.CrossrefPubMed Google Scholar
53 Garnett MJ , EdelmanEJ, HeidornSJ, et al.Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature2012;483:570–575.CrossrefPubMed Google Scholar
54 Shoemaker RH , MonksA, AlleyMC, et al.Development of human tumor cell line panels for use in disease-oriented drug screening. Prog Clin Biol Res1988;276:265–286.PubMed Google Scholar
55 Weinstein JN , MyersTG, O'ConnorPM, et al.An information-intensive approach to the molecular pharmacology of cancer. Science1997;275:343–349.CrossrefPubMed Google Scholar
56 Kwak EL , BangYJ, CamidgeDR, et al.Anaplastic lymphoma kinase inhibition in non–small-cell lung cancer. N Engl J Med2010;363:1693–1703. Google Scholar
57 McDermott U , SettlemanJ. Personalized cancer therapy with selective kinase inhibitors: an emerging paradigm in medical oncology. J Clin Oncol2009;27:5650–5659.CrossrefPubMed Google Scholar
58 McDermott U , SharmaSV, DowellL, et al.Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci2007;104:19936–19941.CrossrefPubMed Google Scholar
59 No authors listed. Genomics of Drug Sensitivity in Cancer Database. http://www.cancerRxgene.org (date last accessed 7 October 2013). Google Scholar
60 Yang W , SoaresJ, GreningerP, et al.Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res2012;41(Database issue):955–961.CrossrefPubMed Google Scholar
61 Heinrich MC , CorlessCL, DemetriGD, et al.Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol2003;21:4342–4349.CrossrefPubMed Google Scholar
62 Barretina J , CaponigroG, StranskyN, et al.The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature2012;483:603–607.CrossrefPubMed Google Scholar
63 No authors listed. Cancer Cell Line Encyclopedia. http://www.broadinstitute.org/ccle/home (date last accessed 7 October 2013). Google Scholar
64 No authors listed. Sanger Cancer Cell Line Project. http://cancer.sanger.ac.uk/cancergenome/projects/cell_lines/ (date last accessed 7 October 2013). Google Scholar
